










- シンクタンクならニッセイ基礎研究所 >
- 社会保障制度 >
- 医療保険制度 >
- 治験の概要-臨床試験の現状 (前編)
治験の概要-臨床試験の現状 (前編)

保険研究部 主席研究員 兼 気候変動リサーチセンター チーフ気候変動アナリスト 兼 ヘルスケアリサーチセンター 主席研究員 篠原 拓也


文字サイズ
- 小
- 中
- 大
4――各フェーズの試験内容
1|第Ⅰ相試験は、被験薬の安全性や、吸収・代謝・分布・排泄を調査する
第Ⅰ相試験は、少人数の健康な成人(通常は男性)を対象に、被験薬の安全性や、吸収・分布・代謝・排泄16を調査する。一般的には、まず、単回投与用量漸増試験(SAD)が行われ、その後、反復投与用量漸増試験(MAD)が行われる。
16 吸収(Absorption)、分布(Distribution)、代謝(Metabolism)、排泄(Excretion)の頭文字をとって、ADME(アドメ)と呼ばれる。ADMEの速度過程は、薬物動態と呼ばれ、医薬品開発の重要な要素とされる。
SADは、安全用量の絞り込み、体内動態の把握を目的に行われる。
まず、健康な人を8名ごとなどの組(「コホート」という)に分ける。非臨床試験での動物への投与結果などをもとに、各コホートに異なる用量が設定される。設定された用量の低いコホートから、たとえば6名には被験薬、2名にはプラセボが二重マスク法で投与されて、それぞれデータが収集される。このコホートで安全性が確認された場合は、医師の判断のもとで、一段階用量の高いコホートへの投与に移る(「コホート移行」という)。コホート移行の障害となる有害事象が発生するか、もしくは、設定されていた最高用量のコホートへの投与が終了した場合、SADは終了となる。
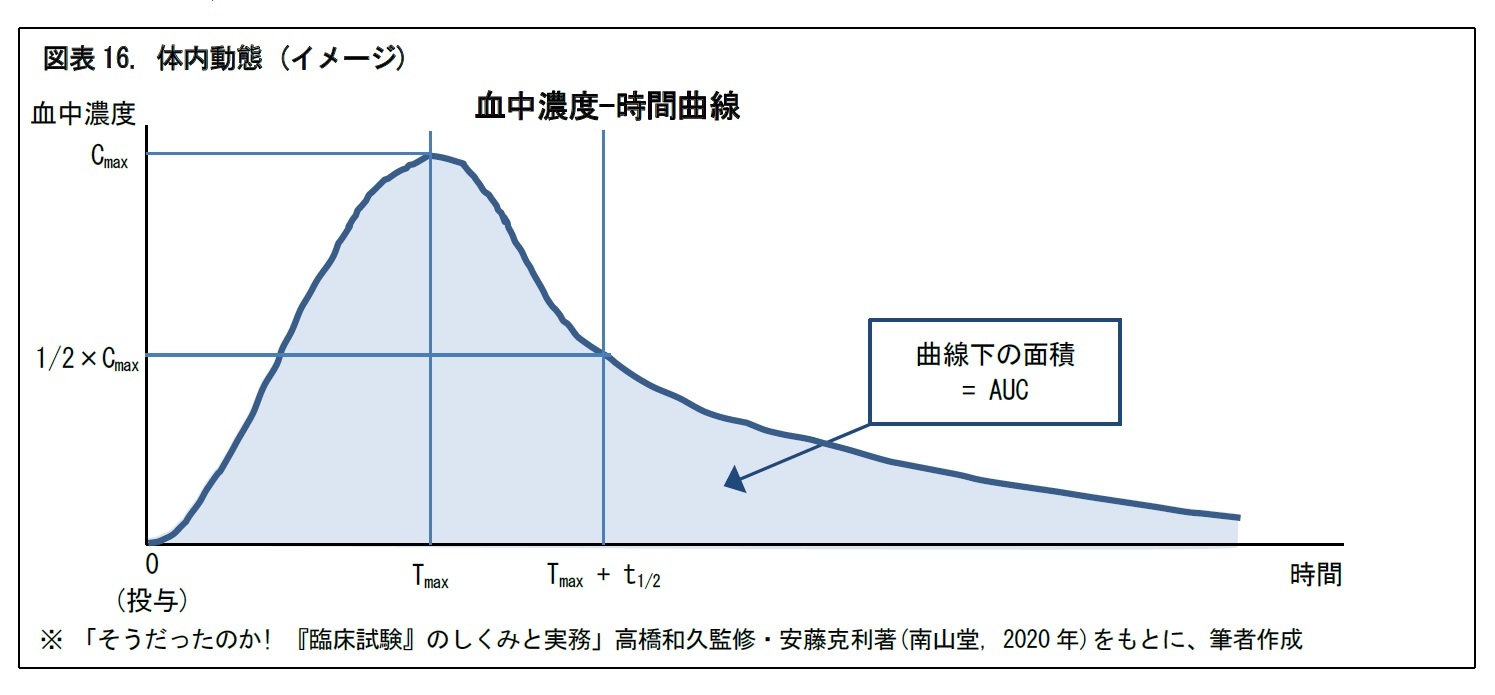
一般に、つぎの図のように、医薬品は剤形によって、投与後の時間経過に応じた血中濃度の推移が異なる。そこで、血中濃度を確認するために、投与後の所定時間ごとに、何回か被験者の採血が行われる。併せて、有害事象の発現の有無や、発現した場合の重症度の確認・評価も行われる。
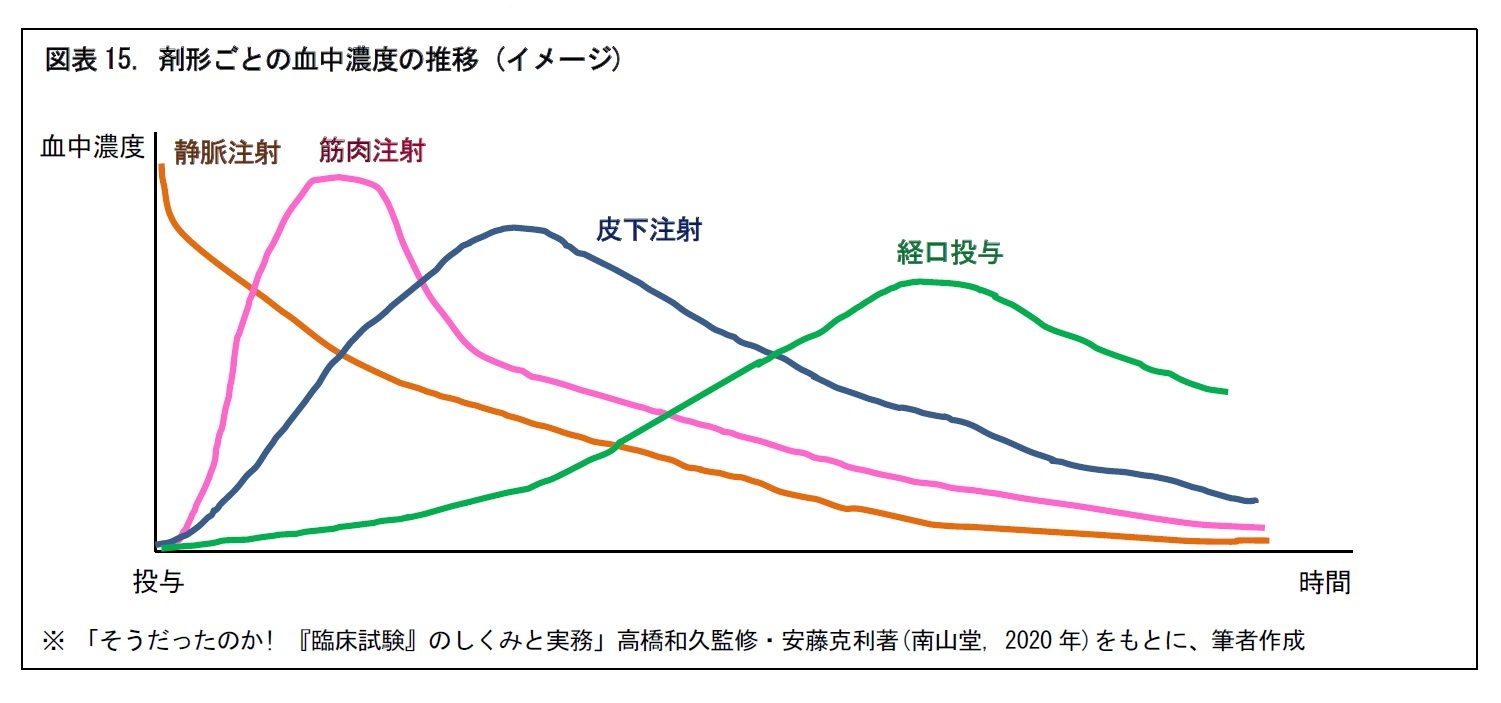
また、日本人と外国人で、t1/2などの血中動態が異なる場合がある。このような場合には、日本からの国際共同治験への参加が困難となることもある。
MADは、反復投与時の安全性の確認、体内動態の把握を目的に行われる。
SADと同様に、健康な人をコホートに分ける。各コホートに異なる用量が設定される。用量の低いコホートから投与を開始し、コホート移行を進める。コホート移行の障害となる有害事象が発生するか、もしくは、設定されていた最高用量のコホートへの投与が終了した場合、MADは終了となる。
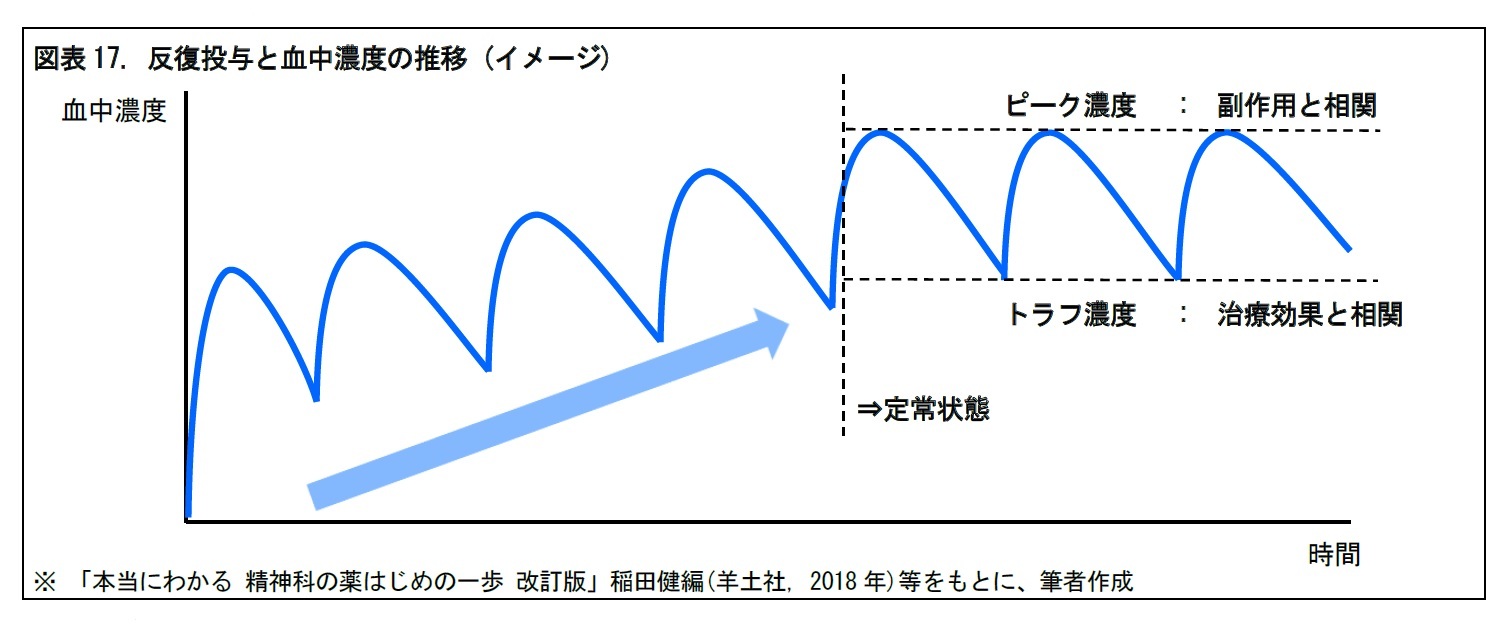
体内動態については、血中濃度が直近の最高値から半減した時に投与を繰り返すことで、血中濃度が有効血中濃度内で推移するか(定常状態になるか)を確認する。反復投与することが多い精神疾患系の薬剤等では、定常状態に達した場合、ピーク濃度(定常状態での最高濃度)は副作用と相関し、トラフ濃度(同最低濃度)は治療効果と相関するといわれる。
なお、SADで線形性が確認されていても、反復投与により蓄積・飽和が起こると、血中濃度が急上昇して線形性が崩れることがあるため、注意が必要とされる。
第I相試験で安全性が確認された用量の範囲内で、少数の患者に対して、被験薬が初めて投与される。第II相試験は、前期試験(P2a)と後期試験(P2b)に分けられる。第II相試験は、用量設定などを探索する試験であるため、「探索的試験」と呼ばれる。
(1) 前期試験(P2a)
前期試験では、被験薬の安全性・有効性・薬物動態をみる。第I相試験と同様に、低用量の投与から開始して、順次、用量を増やしていく。P2a試験では、POCが確認される。POCは、医薬品の開発の妥当性をあらわすもので、新薬開発プロセスのマイルストーンと位置づけられている。
(2) 後期試験(P2b)
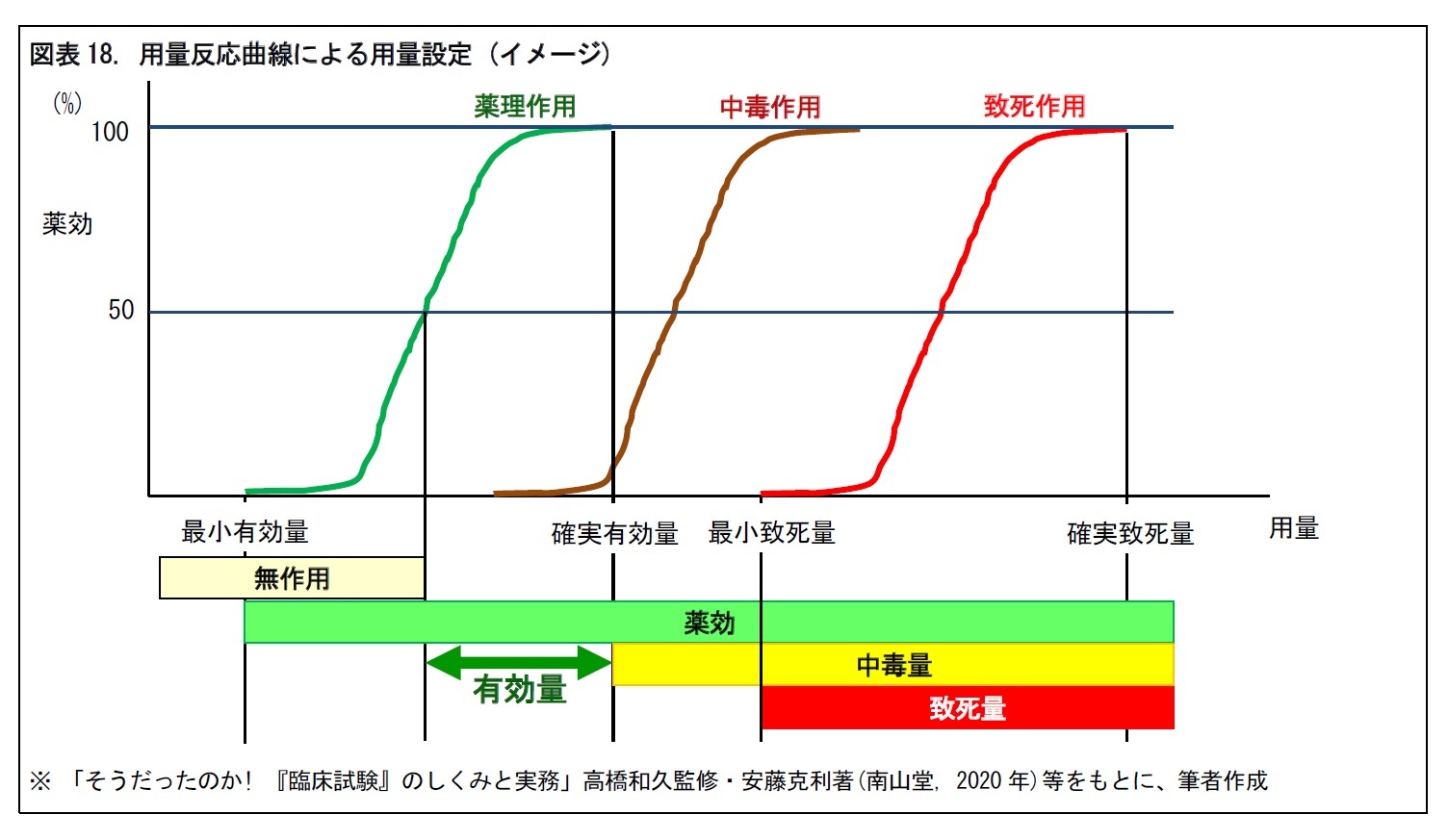
後期試験では、薬物動態・適応症の明確化や第III相試験の用量設定を行う。用量設定では、有効性を得るためには、どの程度の投与量が必要かという、「用量設定試験」が行われる。この試験を通じて、有効性と安全性が両立する「有効量」を明らかにする。
抗がん剤の場合は、第I相試験で患者に投与することで、すでにPOCが確認されていることが多い。このため、第II相試験は、薬物動態と有害事象の関連性の確認などが中心となる。一般に、対照群を置かずに被験者すべてに被験薬を投与する「シングルアーム試験」が行われる。シングルアーム試験では、個々の症例について、改善、状態維持、進行といった状態変化を評価する。具体的には、4パターンに分けて、完全奏功と部分奏功の患者割合の合計(「奏効率」といわれる)で評価する。

第III相試験では、第II相試験で決定された投与量の有効性・安全性について、ランダム化比較試験による検証が行われる。このため、第III相試験は、「検証的試験」と呼ばれる。
通常、対照群には、プラセボが投与される。すでに標準治療が確立されている場合には、その標準治療が行われることもある。その場合、標準治療と比べて、被験薬の有効性が劣っていないことを検証する「非劣性試験」が行われることとなる。
5|複数の相を併用する場合もある
医薬品開発は、第I相~第III相の試験を順番に行うことが基本とされる。しかし、そのためには、多くの患者を被験者として登録することが必要となる。3つのフェーズ(相)の試験を、別々に実施しようとすると、希少疾患や、有効な治療法や薬剤がない疾患領域での医療ニーズ(Unmet Medical Needs, UMN)に応える医薬品の開発は困難となる。
また、抗がん剤のように、第I相試験から患者を被験者として投与するケースもある。必ずしも、3つの段階で、臨床試験を進めることが必要とされる訳ではなく、承認申請時に、被験薬としての安全性・有効性を示すデータが揃っていればよいといえる。
そこで、対象となる疾患や医薬品の特性に準じて、第I相と第II相を併用する「P1/2試験」や、第II相と第III相を併用する「P2/3試験」により、費用や時間の抑制を目指す場合もある。このような臨床試験のデザインが、医薬品開発の成否を分けるケースもあるため、臨床試験においては、そのデザイン設計が重要といえる。
5――ワクチンの治験
1|ワクチンには安全性が強く求められる
感染症に対するワクチンは、健康な人に向けて、予防の目的で投与される。もしワクチンを投与したことで、健康な人が病気になってしまうようなら、大問題となりかねない。
このため、ワクチン開発においては、治験により、有効性とともに安全性の確認が強く行われる。治験は、法令に定める基準やガイドライン17に従って行われる。そのガイドラインによれば、事前に大学などの審査委員会の審査・承認を受けること、被験者のインフォームド・コンセント(十分な説明に基づく同意)を得ること、などとされている。
17 「『感染症予防ワクチンの臨床試験ガイドライン』について」(厚生労働省, 薬食審査発0527 第5号, 平成22年5月27 日)など。
ワクチンの治験は、治療薬の治験と同様、3つのフェーズで行われることが一般的とされている。
(1) 第I相試験
第I相試験は、通常、少人数の健康な成人を被験者として、小規模な試験として行われる。ワクチンの有効性と安全性に関する、予備的な探索を行うことが目的となる。
(2) 第II相試験
第II相試験は、健康な人を被験者として、ワクチンの使用方法などに関する試験として行われることが一般的。被験者に、未成年者や高齢者を含むこともある。ワクチンの接種量、接種スケジュール、接種経路を明確にすることが、主な目的となる。
(3) 第III相試験
第III相試験は、大規模な集団において、有効性と安全性のデータを得ることが目的となる。通常、投与される被験者にも投与する医師にもわからないよう、ランダムにワクチンまたはプラセボを割り当てて投与し、その効果を比較することで有効性をテストする。すなわち、二重マスクでのランダム化比較試験として行われる。
第III相試験は、数千人~数万人規模の集団を被験者とすることもあり、研究開発費の多くが、ここで費やされるといわれる。開発が成功して実用化できるか、それとも失敗に終わるか、まさにワクチン開発の最大のヤマ場といえる。
3|有効性は基本的に発症予防効果でみる
では、有効性の確認は、どのように行われるのだろうか。臨床試験のガイドラインによると、基本的には、「発症予防効果」をみることが望ましい、とされている。ただし、疾患の発生頻度が非常に低い場合等は、発症予防効果をみることが困難な場合が多い。このような場合には、発症予防との相関性が確立されている抗体価などの代替指標を評価することが適切とされる。また、すでに実用化されたワクチンがある場合には、そのワクチンと有効性を比較する「非劣性試験」を行う場合もある。
発症予防効果は、「ワクチンを打たなかった場合と比べて、どれだけ発症する患者を減らせたか」という指標で表される。たとえば、ワクチン本剤とプラセボをそれぞれ100人ずつ被験者に投与したところ、しばらくして、本剤を投与された人からは20人、プラセボを投与された人からは50人が発症したとする。この場合、ワクチンの効果により30人(=50人-20人)の発症が予防できたとみられる。したがって、発症予防効果は、60%(=30人÷50人)となる。
感染症の種類によって、発症予防効果は異なる。たとえば、予防接種の効果が一生涯続くとされる、はしかの場合、発症予防効果は95%以上といわれている18。
一方、季節性インフルエンザでは予防接種を受けても、その効果は数ヵ月間に限られる。ある研究によれば、発症予防効果は65歳以上の健常な高齢者について約45%であったと報告されている19。
このように、ワクチンの効果は100%ではない。たとえワクチンを打っても、感染や発症をしないとは言い切れないことになる。しかし、多くの人がワクチンを打てば、感染者の数を減らすことができ、その結果、感染拡大が抑えられる。つまり、ワクチンによって「集団免疫」が働く効果がある。この集団免疫を効かせるために、早期のワクチン開発、ワクチン接種が望まれることとなる。
18 「麻疹の現状と今後の麻疹対策について」(国立感染症研究所 感染症情報センター, 平成14 年10 月)より。
19 「医療関係者のためのワクチンガイドライン第2版」(一般社団法人日本環境感染学会, 環境感染誌Vol. 29 suppl. III, 2014年) より。
安全性の評価は、投与されたすべての被験者に対して、「有害事象」を収集する形で行われる。
ワクチンの場合、体外の物質が化学作用を起こすことよりも、体内で免疫学的に起こる反応が問題となることが多い。そこで、治療薬の「副作用」とは区別して、「副反応」という用語が使われる。
副反応には、予防接種をした部位が腫れたり、赤みを帯びたり、ズキズキ痛んだりする局所反応と、発熱やリンパ節が腫れるなどの全身反応がある。局所反応や全身反応の多くは、投与後数日以内に発現するとされる。
特に、重篤な有害事象として、死亡・障害やそのおそれのある症例、後世代における先天性の疾患・異常などがあげられる。こうした重篤な有害事象に対しては、詳細な報告書を作成するとともに、報告後も十分にモニタリングを行う必要があるとされている。
有害事象を収集する期間は、微生物の全体または一部を感染しないように無毒化して免疫を獲得する不活化ワクチンの場合、投与後2週間。ウイルスなどの原因微生物を発症しない程度に弱毒化したうえで使用する生ワクチンの場合、投与後4週間が目安とされている。電話連絡により確認したり、被験者が受診する際に日誌を回収したりして、収集される。
このように、ワクチンの場合は、安全性に対する評価がとても重視される。発症予防効果がいくら高くても、副反応のリスクが大きければ、開発はストップされる。つまり、ワクチンの開発はとても難しいということになる。
(参考) 新コロナウイルス感染症ワクチンの治験
新型コロナウイルス感染症について、日本では、現在(2021年7月)までに3つのワクチンが特例承認 * のうえ、実用化されている。治験で確認された、主な有効性と安全性についてまとめておく。
(1) ファイザー社のワクチン「コミナティ筋注」 (2021年2月14日特例承認)
1) 有効性
海外の第II/III相試験において、治験薬接種前から 2回目接種後7日以前にSARS-CoV-2感染歴がない被験者における発症予防効果(ワクチン有効率)は、95.0% [95%信用区間:90.3~97.6%]
2) 安全性
海外の第I/II/III相試験では、重篤な有害事象(SAE)が本剤群126/21,621例(0.6%)、プラセボ群111/21,631例(0.5%)に認められた。死亡例は、本剤群2例、プラセボ群4例に認められ、いずれも治験薬との因果関係は否定された。なお、国内の第I/II相試験では、死亡及びSAEは認められなかった。
(2) アストラゼネカ社のワクチン「バキスゼブリア筋注」 (2021年5月21日特例承認)
1) 有効性
海外の第II/III相試験において、主解析対象集団**の2回目の接種後15日以降に発現した初発のCOVID-19発症に対する発症予防効果(ワクチン有効率)は、70.42% [95%信用区間:54.83~ 80.63%]
2) 安全性
海外の第I/II/III相試験では、SAEが本剤群108/12,282例(0.9%)、対照群127/11,962例(1.1%)に認められた。死亡例は、本剤群2例、対照群5例に認められ、いずれも治験薬との因果関係は否定された。国内の第Ⅰ/Ⅱ相試験では、本剤群において死亡及びSAEは認められなかった。
(3) 武田/モデルナ社のワクチン「COVID-19ワクチンモデルナ筋注」 (2021年5月21日特例承認)
1) 有効性
海外の第III相試験において、治験薬接種前から2回目接種後14日以前にSARS-CoV-2感染歴がない被験者における発症予防効果(ワクチン有効率)は、94.1% [95%信頼区間:89.3%~96.8%]
2) 安全性
海外の第III相試験では、SAEが本剤群147/15,185例(1.0%)、プラセボ群151/15,166例(1.0%)に認められた。死亡例は、本剤群6例、対照群7例に認められ、いずれも治験薬との因果関係は否定された。国内の第I/II相試験では、本剤群において死亡及びSAEは認められなかった。
* 医薬品医療機器等法(薬機法)第14条の3第1項の規定に基づき、(1)疾病の蔓延防止等のために緊急の使用が必要、(2)当該医薬品の使用以外に適切な方法がない、(3)海外で販売等が認められている、という要件を満たす医薬品につき、承認申請資料のうち臨床試験以外のものを承認後の提出としても良い等として、特例的な承認をする制度。
** 2回とも標準用量、もしくは、1回目低用量・2回目標準用量として、2回接種した集団
※「今後の新型コロナワクチンの接種について」(第19回厚生科学審議会予防接種・ワクチン分科会, 資料1, 2021年2月15日)および「新型コロナワクチンの接種について」(第21回厚生科学審議会予防接種・ワクチン分科会, 資料1, 2021年5月21日)をもとに、筆者が表現を一部改変
(2021年07月29日「基礎研レポート」)

 各種レポート配信をメールでお知らせ。読み逃しを防ぎます!
各種レポート配信をメールでお知らせ。読み逃しを防ぎます!
保険研究部 主席研究員 兼 気候変動リサーチセンター チーフ気候変動アナリスト 兼 ヘルスケアリサーチセンター 主席研究員
篠原 拓也 (しのはら たくや)
研究・専門分野
保険商品・計理、共済計理人・コンサルティング業務
03-3512-1823
- 【職歴】
1992年 日本生命保険相互会社入社
2014年 ニッセイ基礎研究所へ
【加入団体等】
・日本アクチュアリー会 正会員
篠原 拓也のレポート
| 日付 | タイトル | 執筆者 | 媒体 |
|---|---|---|---|
| 2025/05/27 | 気候指数 2024年データへの更新-日本の気候の極端さは1971年以降の最高水準を大幅に更新 | 篠原 拓也 | 基礎研レポート |
| 2025/05/20 | 「次元の呪い」への対処-モデルの精度を上げるにはどうしたらよいか? | 篠原 拓也 | 研究員の眼 |
| 2025/05/13 | チェス盤を用いた伝心-愛情と計算力があれば心は通じる? | 篠原 拓也 | 研究員の眼 |
| 2025/05/09 | 国民負担率 24年度45.8%の見込み-高齢化を背景に、欧州諸国との差は徐々に縮小 | 篠原 拓也 | 基礎研マンスリー |






新着記事
-
2025年10月24日
消費者物価(全国25年9月)-コアCPI上昇率は拡大したが、先行きは鈍化へ -
2025年10月24日
保険業界が注目する“やせ薬”?-GLP-1は死亡率改善効果をもたらすのか -
2025年10月23日
御社のブランドは澄んでますか?-ブランド透明性が生みだす信頼とサステナビリティ開示のあり方(1) -
2025年10月23日
EIOPAがソルベンシーIIのレビューに関する技術基準とガイドラインのセットの新たな協議を開始等 -
2025年10月23日
中国:25年7~9月期GDPの評価-秋風索莫の気配が漂う中国経済。内需の悪化により成長率は減速
レポート紹介
-
研究領域
-
経済
-
金融・為替
-
資産運用・資産形成
-
年金
-
社会保障制度
-
保険
-
不動産
-
経営・ビジネス
-
暮らし
-
ジェロントロジー(高齢社会総合研究)
-
医療・介護・健康・ヘルスケア
-
政策提言
-
-
注目テーマ・キーワード
-
統計・指標・重要イベント
-
媒体
- アクセスランキング
お知らせ
-
2025年07月01日
News Release
-
2025年06月06日
News Release
-
2025年04月02日
News Release
【治験の概要-臨床試験の現状 (前編)】【シンクタンク】ニッセイ基礎研究所は、保険・年金・社会保障、経済・金融・不動産、暮らし・高齢社会、経営・ビジネスなどの各専門領域の研究員を抱え、様々な情報提供を行っています。
治験の概要-臨床試験の現状 (前編)のレポート Topへ- 新型コロナウイルス
- ウィズコロナ・アフターコロナ
- 生成AI・AI
- IoT
- デジタルトランスフォーメーション(DX)
- フィンテック(FinTech)
- キャッシュレス
- デジタル通貨
- デジタルプラットフォーム
- マイナンバー
- MaaS、CASE
- SDGs
- ESG
- 気候変動
- カーボンニュートラル・脱炭素社会
- サステナビリティ
- ウェルビーイング
- 生物多様性
- イデコ(iDeCo)
- 新NISA・NISA
- 日本銀行
- 人手不足・人材不足
- 働き方改革
- 人的資本経営
- 従業員エンゲージメント
- テレワーク・在宅勤務
- ダイバーシティ(多様性)社会
- 外国人雇用・就労
- 労働政策
- 地域包括ケア・地域共生社会
- 認知症
- 金融(ファイナンシャル)ジェロントロジー
- 全世代型社会保障
- 社会保障・税改革
- 医療・介護制度改革
- 健康寿命
- 健康経営
- 格差・貧困
- 世代間格差
- パワーカップル
- 未婚化
- プレコンセプションケア
- 少子高齢化
- 東京一極集中
- インバウンド
- シェアリングエコノミー
- Z世代・α世代
- エンタメ
- オフィスレントインデックス
- 生命保険事業概況
- 米中貿易摩擦
- 米国
- 中国
- 欧州
- アジア・新興国
- 韓国
- ASEAN
- 統計
- 確定拠出年金
- 企業型DC
- 資産所得倍増プラン
- 金融リテラシー
- 住宅リテラシー
- 年金制度改革
- インド
- 経済安全保障
- 供給網(サプライ・チェーン)
- 消費者物価指数(CPI)│日本
- 雇用統計│日本
- 鉱工業生産指数│日本
- 貿易統計│日本
- 法人企業統計│日本
- QE速報・予測
- 日銀金融政策決定会合
- 日銀短観│日本
- 資金循環統計│日本
- 景気ウォッチャー調査│日本
- 地域経済報告(さくらレポート)
- マネタリーベース│日本
- GDP等│米国
- FOMC(連邦公開市場委員会)│米国
- 住宅販売・着工│米国
- 雇用統計│米国
- 米個人所得・支出|米国
- ECB政策理事会│欧州
- ユーロ圏消費者物価指数
- ユーロ圏GDP
- ユーロ圏失業率
- 英国雇用関連統計
- 英国金融政策
- 英国GDP
- 将来人口推計
- 人口動態統計
- 宿泊旅行統計
- 中国GDP
- インドGDP
- タイGDP
- マレーシアGDP
- フィリピンGDP
- インドネシアGDP
- ベトナムGDP
- ロシアGDP
- ブラジルGDP
- IMF世界経済見通し
- 企業物価指数
- インド消費者物価
Copyright © NLI Research Institute. All rights reserved.