










- シンクタンクならニッセイ基礎研究所 >
- 社会保障制度 >
- 医療保険制度 >
- 治験の概要-臨床試験の現状 (前編)
治験の概要-臨床試験の現状 (前編)

保険研究部 主席研究員 兼 気候変動リサーチセンター チーフ気候変動アナリスト 兼 ヘルスケアリサーチセンター 主席研究員 篠原 拓也


文字サイズ
- 小
- 中
- 大
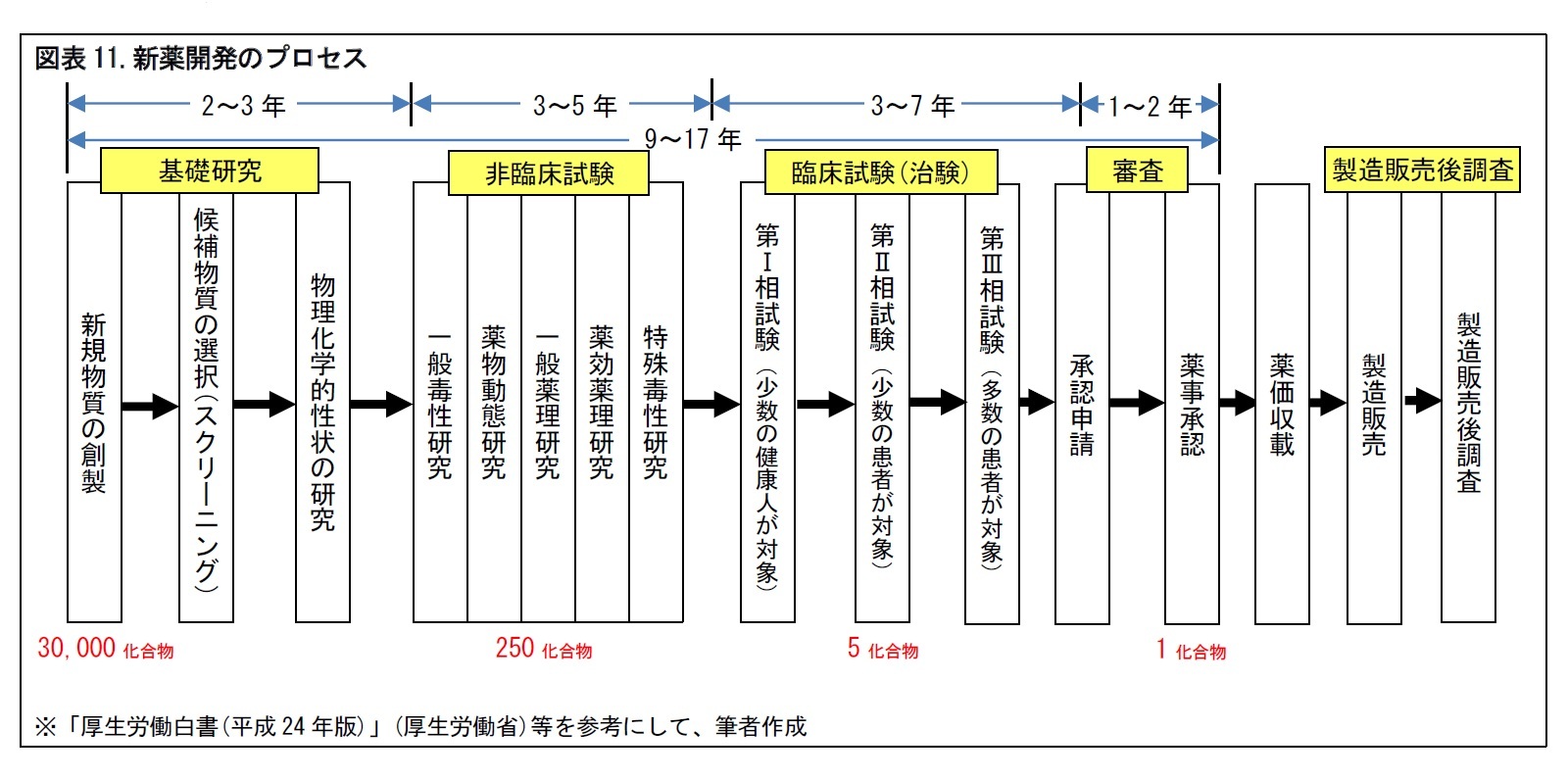
2――新薬の開発プロセス
1|治験は、新薬開発のカギ
新薬を開発するプロセスをみていこう。新薬開発は、大まかに、基礎研究、非臨床試験、臨床試験(治験)、審査、製造販売後調査の順に進められる。
(参考) 新薬の特許
一般に、工業製品の特許権存続期間は、出願から原則20年とされている。このルールは、医薬品にも適用され、通常、新薬開発中に出願される。ただし、出願後でも開発中は特許権の行使はできない。医薬品は開発に長期間を要するため、多くの場合、5年間の特許期間延長が認められる。
特許権存続期間や再審査期間中は、他の医薬品メーカーは、後発医薬品を製造販売することはできない。このため、新薬を開発した医薬品メーカーは、独占販売が可能となる。
2|治験では、第III相試験が開発のヤマ場
通常、治験は、第I相、第II相、第III相の3つの試験の段階を踏んで実施される。必要に応じて、薬物動態や薬力学的作用をみるための臨床薬理試験が行われる。
(1) 第I相試験
少人数の健康な成人(通常は男性)を対象8に、安全性とヒトにおける薬物動態をみる。まず、単回投与用量漸増試験(Single Ascending Dose, SAD)を行い、安全性に問題がないことを確認したうえで、反復投与用量漸増試験(Multiple Ascending Dose, MAD)を行うことが一般的とされる。
ヒトに対して初めて投与する試験(First In Human試験, FIH)では、非臨床試験の毒性研究で得られた無毒性量をもとに、低用量から投与を開始するなど、慎重に試験が行われる。
8 抗がん剤のように毒性の強い薬物の場合は、第Ⅰ相試験から、患者を対象とすることもある。
少人数の患者を対象に、複数の用量で有効性と安全性をみてゆき、それをもとに第III相試験の用法・用量を設定する。新しい作用機序を見込む医薬品の開発で、その有効性を見極める試験は、POC(Proof Of Concept, 「ポック」と発音)試験と呼ばれる。第II相試験は、POC試験と位置づけられることが多い。POCを確認することは、新薬開発の重要なマイルストーンの1つとなるため、第II相試験の重要性は高い9。
また、海外ですでに行われた第III相試験の結果を、日本人の結果に外挿するために、海外で行われた第II相試験と同じ設定のもとで、日本で第II相試験を行うことがある。これは、「ブリッジング試験」と呼ばれ、開発期間の短縮や、開発費用の抑制を目的として行われる。ブリッジング試験では、設定の細部に留意することが求められる。
9 抗がん剤の場合は、第I相試験で、POCがすでに確認されていることも多い。
多数の患者を対象に、至適用法・用量10での有効性、安全性を検証する。通常は、二重マスクでのランダム化比較試験として行い、被験薬11がプラセボ群に比べて有効であること(優越性)、もしくは既承認薬に対して有効性が劣っていないこと(非劣性)を検証する。
試験に参加してもらう患者数として、被験薬の有効性を統計学的に示すために必要となる症例数が設定される。被験薬が慢性疾患に対する医薬品の場合、長期間投与した場合の安全性や有効性を検証するために、長期投与試験も行われる。
なお、近年は、複数の国で共通の治験実施計画書(PC)を用いて実施する「国際共同治験」が増えている12。治験においては、各段階で、独立行政法人医薬品医療機器総合機構(PMDA)と治験相談を重ねつつ、当局との合意のもとで進めることが、一般的とされる。
10 「至適」は、医学や医療の分野で用いられる表現で、「最適な」といった意味合いを持つ。
11 本稿では、「被験薬」は、治験の対象とされる薬物をいう。また、「治験薬」は、被験薬と対照薬を表す。
12 詳しくは、第5章を参照。
治験が終了し、申請資料がまとまると、厚生労働大臣の薬事承認を取得するための申請が行われる。通常、PMDAが実質的な審査を行うため、医薬品メーカーはPMDAに対して承認申請を行う。
PMDAは、申請資料の信頼性を確認しつつ、各領域の専門家からなる審査チームを構成して審査を進める。必要に応じて、外部専門家の意見も聴き、審査報告書を作成する。できあがった審査報告書は、PMDAから厚生労働省に提出される。
厚生労働省では、その審査報告書をもとに、薬事・食品衛生審議会で審議を進める。審議会の答申を踏まえて、最終的に、大臣が承認を行う。
4|製造販売後調査の結果をもとに再審査申請が行われる
医薬品は、承認を受けて販売された後にも、安全性確認のために「製造販売後調査」が行われる。実際に臨床医療で使用した医薬品について、副作用の発生状況などの安全性情報が収集される。
通常、薬事承認の際には、再審査期間が設定される。医薬品メーカーは、再審査期間の満了後に、製造販売後調査の結果をもとに、再審査申請を行う必要がある。
製造販売後調査は、「医薬品製品販売後安全管理の基準」(Good Vigilance Practice, GVP)と、「医薬品製造販売後調査・試験の実施の基準」(Good Post-marketing Study Practice, GPSP)という2つのルールに従って行われる。市販直後調査、使用成績調査、製造販売後データベース調査、製造販売後臨床試験に分けられる。
(1) 市販直後調査
販売後6カ月間、GVPのもとで、医療機関に対して確実な情報提供や注意喚起を行うこととあわせて、副作用などの情報を収集する。
(2) 使用成績調査
新規承認された医薬品に対しては、GPSPのもとで、投与された全症例について、副作用の発生状況や、有効性・安全性に関する情報を検出して確認することが求められる。使用成績調査は、「一般使用成績調査」、「特定使用成績調査」、「使用成績比較調査」に分けられる13。このうち、特定使用成績調査は、 特定の患者(小児、高齢者、妊産婦、腎・肝機能障害を有する患者)や、長期間に医薬品を使用する患者を対象に行われる調査をいう。
13 一般使用成績調査は、医薬品を使用する者の条件を定めることなく行う調査。使用成績比較調査は、特定の医薬品を使用する者の情報と当該医薬品を使用しない者の情報とを比較することによって行う調査。
医療情報データベースを用いて、医薬品の副作用による疾病等の種類別の発生状況などを検出して確認する。調査を開始する前に、同データベースの信頼性を担保しておくことが必要となる。もし、信頼性を担保できないことが判明した場合は、調査計画を練り直すことが必要となる。
(4) 製造販売後臨床試験
希少疾患に対する医薬品などで、登録症例数が少ない場合、承認時に、製造販売後臨床試験(第IV相試験)の実施が課される場合がある。その場合、臨床試験を行い、その結果をもとに再審査が行われる。再審査の結果、安全性や有効性に問題があると判断されると、承認が取り消されることもある。製造販売後臨床試験で取得するデータは、医薬品メーカーが設定する。第I相~第III相試験と同様に、被験者となる患者の同意や、治験審査委員会(IRB)の承認手続きが必要となる。
市販後の各調査で得られた結果は、「安全性定期報告書」にまとめられる。厚生労働大臣の指定した日(医薬品の販売が認められた日など)から2年間は6ヵ月ごと、その後は1年ごとに報告することが必要となる。
さらに、これらの定期報告とは別に、医薬品医療機器等法(薬機法)14には、所定の副作用が発生した場合、医薬品メーカーは、(PMDAを通じて)厚生労働大臣に報告する義務があることが、規定されている。
14 法律の正式名称は、「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」。
3――治験のルール
1|被験者の人権保護が科学・社会のための利益に優先
治験を実施する際は、基本的ルールを遵守する必要がある。
厚生労働省が公表しているガイダンスによると、被験者の人権の保護、安全の保持、福祉の向上に対する配慮が最も重要であり、これらは、科学と社会のための利益よりも優先されるべきとされている(③)。また、全ての被験者から、治験参加前に、自由意思によるインフォームド・コンセントを得ることとされている(⑨)。さらに、被験者の身元を明らかにする可能性のある記録は、プライバシーと秘密の保全に配慮して保護すること、なども示されている(⑪)。

治験は、医薬品メーカーや医療機関が勝手に実施できるわけではない。実施するには、行政への届出・報告が必要となる。
(1) 治験届の提出
新薬の治験を実施する前には、厚生労働大臣に治験届を提出する必要がある。新薬を初めて日本人に投与する治験計画については、PMDAによる30日間の調査(「30日調査」)が行われる。届出後30日が経過するまで、医薬品メーカーは、実施医療機関に治験を依頼できない。PMDAは、この30日の間に、被験者の安全性確保の観点から調査を行い、必要があれば治験の中止や変更を指示する。
(2) 副作用の報告
治験中に発生した「重篤な有害事象」(Serious Adverse Event, SAE)のうち、未知の副作用については、PMDAを通じて、厚生労働大臣への報告が必要となる。報告期限は、死亡または死亡のおそれについては7日以内、その他の場合は15日以内とされている。一方、死亡または死亡のおそれのある既知の副作用については、15日以内に報告することとされている。
3|治験は、倫理規範のもと、法令に基づいて行われる
治験は、患者を含むヒトを対象に行われる。そのため、人権保護などの倫理規範のもとで、法令を遵守して行なわなければならない。臨床研究に対して、倫理規範となっているものには、「ニュルンベルク綱領」と「ヘルシンキ宣言」がある。
(1) ニュルンベルク綱領
臨床研究における人権保護の取り組みの原点は、第二次世界大戦にある。大戦中に、ナチス・ドイツが捕虜等に対して行った人体実験や非人道的行為に対する国際軍事裁判が、1946年にドイツのニュルンベルクで開かれた。その判決文のなかで、こうしたことが二度と繰り返されることのないよう、ニュルンベルク綱領の原型が示された。同綱領は、被験者の理解・納得の上での自発的な同意や、被験者による実験の中止など、臨床研究で遵守すべき10項目の基本原則を定めている。

ニュルンベルク綱領に基づいて、1964年にフィンランドのヘルシンキで行われた第18回世界医師会(World Medical Association, WMA)総会で、医学の研究倫理に関する規範が採択された。その後のWMA総会において、9度修正が行われ、現在の内容に至っている15。同綱領には、序文と一般原則を含めて12のテーマで、37の具体内容が示されている。リスクと負担、インフォームド・コンセント、被験者への害に対する補償や治療の提供などが規定されている。

15 現在の内容は、2013年にブラジルのフォルタレザで開催されたWMA総会で修正されたもの。
(3) GCP省令
医薬品の承認申請にあたり、薬機法第14条では、厚生労働省令に定める基準に従って収集・作成された資料を申請書に添付することが規定されている。
治験に関するものについては、その基準は、「医薬品の臨床試験の実施の基準に関する省令」(GCP省令)とされており、治験の準備・管理・実施等に関する法令基準となっている。
(2021年07月29日「基礎研レポート」)

 各種レポート配信をメールでお知らせ。読み逃しを防ぎます!
各種レポート配信をメールでお知らせ。読み逃しを防ぎます!
保険研究部 主席研究員 兼 気候変動リサーチセンター チーフ気候変動アナリスト 兼 ヘルスケアリサーチセンター 主席研究員
篠原 拓也 (しのはら たくや)
研究・専門分野
保険商品・計理、共済計理人・コンサルティング業務
03-3512-1823
- 【職歴】
1992年 日本生命保険相互会社入社
2014年 ニッセイ基礎研究所へ
【加入団体等】
・日本アクチュアリー会 正会員
篠原 拓也のレポート
| 日付 | タイトル | 執筆者 | 媒体 |
|---|---|---|---|
| 2025/05/27 | 気候指数 2024年データへの更新-日本の気候の極端さは1971年以降の最高水準を大幅に更新 | 篠原 拓也 | 基礎研レポート |
| 2025/05/20 | 「次元の呪い」への対処-モデルの精度を上げるにはどうしたらよいか? | 篠原 拓也 | 研究員の眼 |
| 2025/05/13 | チェス盤を用いた伝心-愛情と計算力があれば心は通じる? | 篠原 拓也 | 研究員の眼 |
| 2025/05/09 | 国民負担率 24年度45.8%の見込み-高齢化を背景に、欧州諸国との差は徐々に縮小 | 篠原 拓也 | 基礎研マンスリー |






新着記事
-
2025年11月14日
マレーシアGDP(2025年7-9月期)~内需は底堅く、外需は純輸出が改善 -
2025年11月14日
保険と年金基金における各種リスクと今後の状況(欧州 2025.10)-EIOPAが公表している報告書(2025年10月)の紹介 -
2025年11月14日
中国の不動産関連統計(25年10月)~販売が一段と悪化 -
2025年11月14日
英国GDP(2025年7-9月期)-前期比0.1%で2四半期連続の成長減速 -
2025年11月14日
家計消費の動向(二人以上世帯:~2025年9月)-「メリハリ消費」継続の中、前向きな変化の兆しも
お知らせ
-
2025年07月01日
News Release
-
2025年06月06日
News Release
-
2025年04月02日
News Release
【治験の概要-臨床試験の現状 (前編)】【シンクタンク】ニッセイ基礎研究所は、保険・年金・社会保障、経済・金融・不動産、暮らし・高齢社会、経営・ビジネスなどの各専門領域の研究員を抱え、様々な情報提供を行っています。
治験の概要-臨床試験の現状 (前編)のレポート Topへ- 新型コロナウイルス
- ウィズコロナ・アフターコロナ
- 生成AI・AI
- IoT
- デジタルトランスフォーメーション(DX)
- フィンテック(FinTech)
- キャッシュレス
- デジタル通貨
- デジタルプラットフォーム
- マイナンバー
- MaaS、CASE
- SDGs
- ESG
- 気候変動
- カーボンニュートラル・脱炭素社会
- サステナビリティ
- ウェルビーイング
- 生物多様性
- イデコ(iDeCo)
- 新NISA・NISA
- 日本銀行
- 人手不足・人材不足
- 働き方改革
- 人的資本経営
- 従業員エンゲージメント
- テレワーク・在宅勤務
- ダイバーシティ(多様性)社会
- 外国人雇用・就労
- 労働政策
- 地域包括ケア・地域共生社会
- 認知症
- 金融(ファイナンシャル)ジェロントロジー
- 全世代型社会保障
- 社会保障・税改革
- 医療・介護制度改革
- 健康寿命
- 健康経営
- 格差・貧困
- 世代間格差
- パワーカップル
- 未婚化
- プレコンセプションケア
- 少子高齢化
- 東京一極集中
- インバウンド
- シェアリングエコノミー
- Z世代・α世代
- エンタメ
- オフィスレントインデックス
- 生命保険事業概況
- 米中貿易摩擦
- 米国
- 中国
- 欧州
- アジア・新興国
- 韓国
- ASEAN
- 統計
- 確定拠出年金
- 企業型DC
- 資産所得倍増プラン
- 金融リテラシー
- 住宅リテラシー
- 年金制度改革
- インド
- 経済安全保障
- 供給網(サプライ・チェーン)
- 消費者物価指数(CPI)│日本
- 雇用統計│日本
- 鉱工業生産指数│日本
- 貿易統計│日本
- 法人企業統計│日本
- QE速報・予測
- 日銀金融政策決定会合
- 日銀短観│日本
- 資金循環統計│日本
- 景気ウォッチャー調査│日本
- 地域経済報告(さくらレポート)
- マネタリーベース│日本
- GDP等│米国
- FOMC(連邦公開市場委員会)│米国
- 住宅販売・着工│米国
- 雇用統計│米国
- 米個人所得・支出|米国
- ECB政策理事会│欧州
- ユーロ圏消費者物価指数
- ユーロ圏GDP
- ユーロ圏失業率
- 英国雇用関連統計
- 英国金融政策
- 英国GDP
- 将来人口推計
- 人口動態統計
- 宿泊旅行統計
- 中国GDP
- インドGDP
- タイGDP
- マレーシアGDP
- フィリピンGDP
- インドネシアGDP
- ベトナムGDP
- ロシアGDP
- ブラジルGDP
- IMF世界経済見通し
- 企業物価指数
- インド消費者物価
Copyright © NLI Research Institute. All rights reserved.