










- シンクタンクならニッセイ基礎研究所 >
- 社会保障制度 >
- 医療保険制度 >
- 治験の実務-臨床試験の現状 (後編)
治験の実務-臨床試験の現状 (後編)

保険研究部 主席研究員 兼 気候変動リサーチセンター チーフ気候変動アナリスト 兼 ヘルスケアリサーチセンター 主席研究員 篠原 拓也


文字サイズ
- 小
- 中
- 大
5――医師主導治験
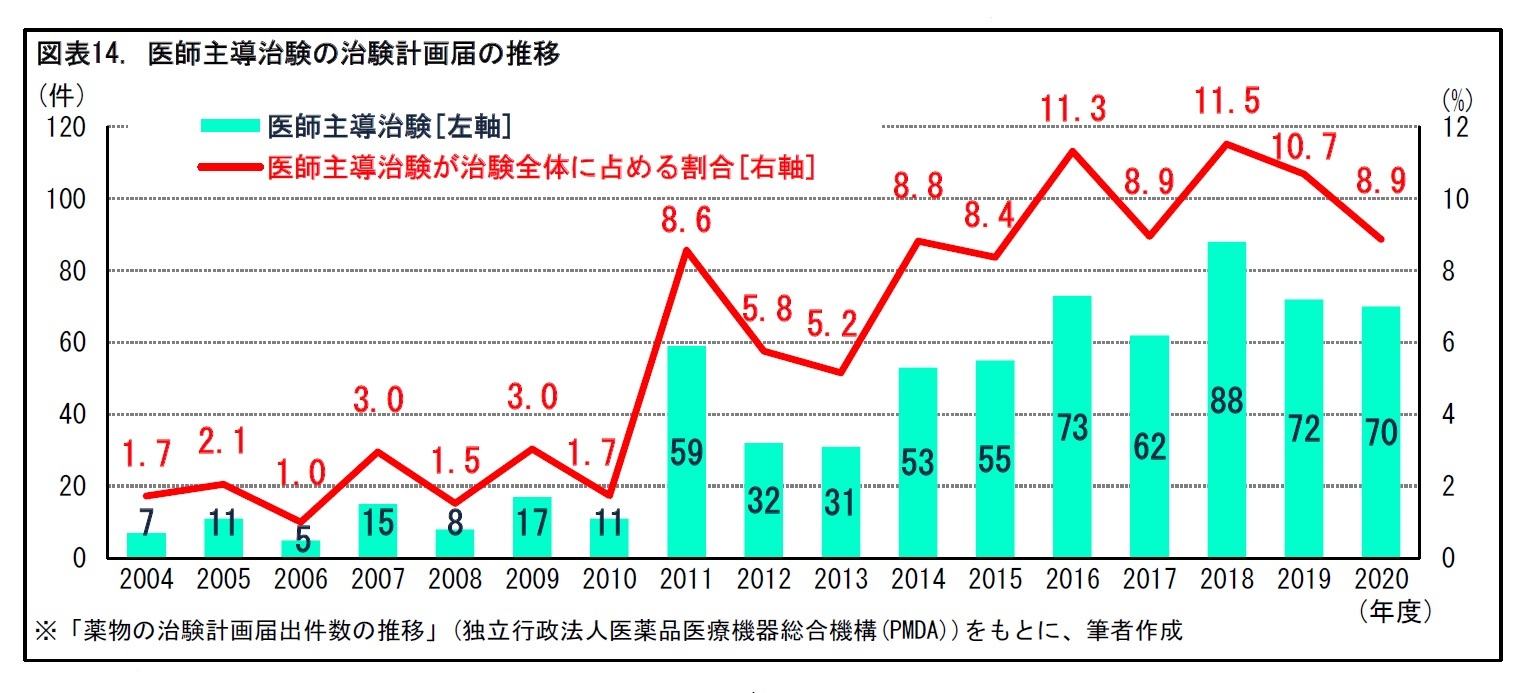
1|医師主導治験が治験全体に占める割合は1割程度
創薬には、医療上の必要性が高くても、経済的な面から、医薬品メーカーが手掛けにくいものがある。特に、小児疾患や希少疾患では、多額の費用がかかる治験で、被験者となる患者を集めにくい。
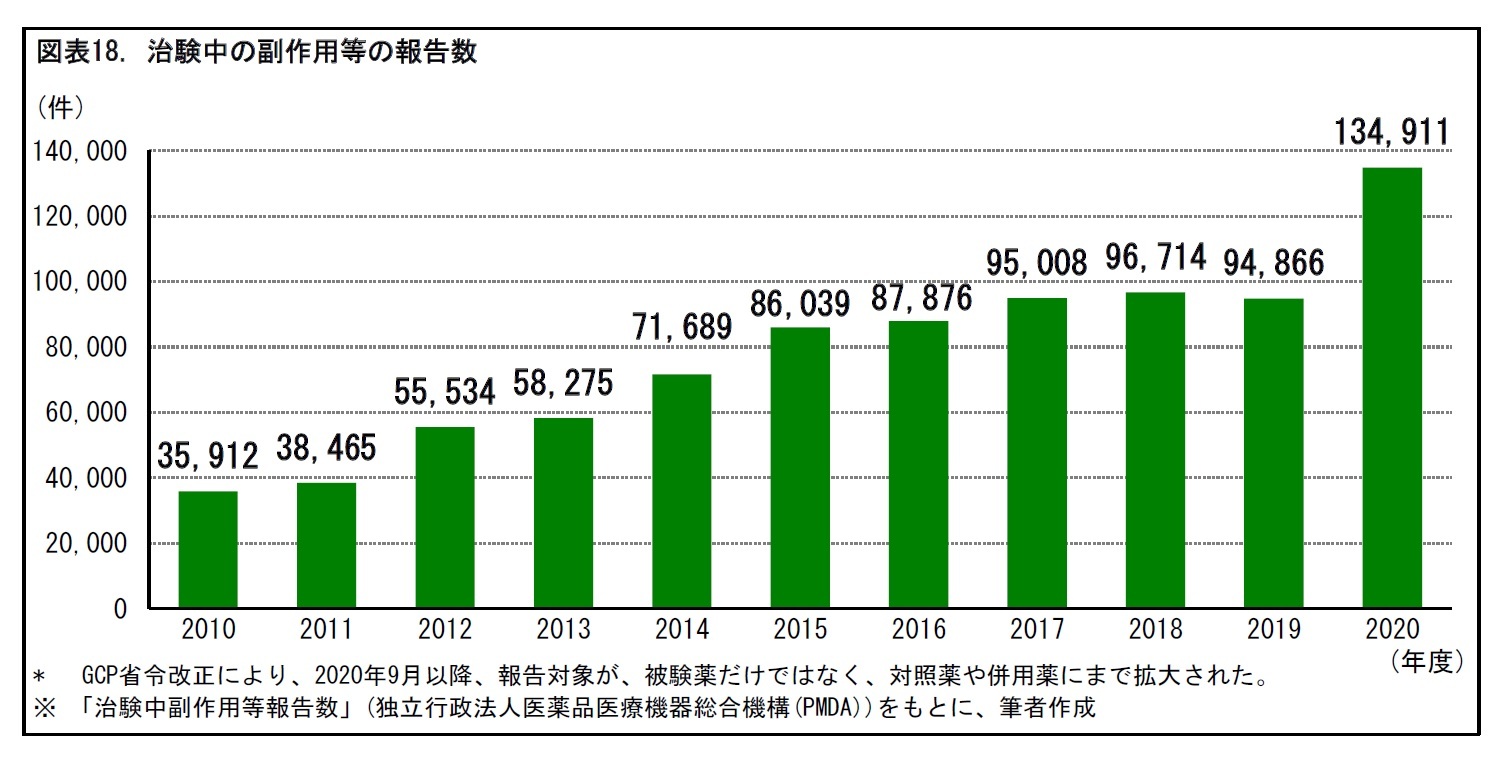
2002年の薬機法改正と、それを受けた2003年のGCP省令改正により、医師主導治験が可能となった。すでに別の効能効果によって承認された医薬品の適応拡大を目的としたり、海外で製造・販売されている医薬品について日本での承認取得を目指したりする場合などに、医師主導治験が行われることがある。医師主導治験は、2010年代に入って増加傾向となっている。2020年度には、70件の医師主導治験の届出があった。だが、治験全体に占める割合は8.9%と、1割程度にとどまっている。
GCPの規定における、「自ら治験を実施する者」とは、厚生労働大臣に治験計画を届け出た治験責任医師とされている。この自ら治験を実施する者が、企業治験での治験依頼者にかわって、治験の準備や管理等の、あらゆる業務を担うこととなる。
ただし、実際問題として、1人の医師だけで治験を実施することは極めて困難であるため、通常は、治験の支援体制がとられる。たとえば、モニタリング、統計解析、総括報告書作成等の業務を、開発業務受託機関(CRO)に委託する事例がみられる。また、大学病院での治験の場合、大学等が医薬品開発を支援する組織として、臨床研究センター等の、アカデミック臨床研究機関(Academic Research Organization, ARO)を設けて、支援するケースもある。
なお、医師主導治験の場合でも、厚生労働省への承認申請は、自ら治験を実施する者(治験責任医師)が行うわけではない。承認申請は、医薬品メーカーが行うこととされている。
また、複数の実施医療機関で、同一の治験実施計画書(PC)にもとづいて、治験を実施する場合がある。このような場合は、あらかじめPCの解釈や、治験の実施手順を実施医療機関の間で統一させておく必要がある。その役割を担うのが、「治験調整医師」である。治験調整医師を支援するために、治験調整事務局が設置されることもある。
3|医師主導治験は、企業治験と異なる点が多い
医師主導治験は、企業治験に比べると、自ら治験を実施する者や、それを支援するCRO等の業務が多くなる。また、つぎのような実務上の相違点もある。
・ 厚生労働大臣への治験計画届の提出が、自ら治験を実施する者の業務となる。そのため、提出の前に、実施医療機関の長から、治験実施の承認を取得しておく必要がある。
・ モニタリングや監査を実施医療機関で行うため、手順書や報告書の第三者による審議が必須となる。その審議のために、治験審査委員会(IRB)を設置することが必要となる。なお、モニタリングや監査を行う際は、治験の実施に従事しない者のなかから選任しなくてはならない。
・ 重篤な有害事象(SAE)が発生した場合、自ら治験を実施する者が、厚生労働大臣への報告を行わなくてはならない。
・ 医師主導治験では、研究費が限られているため、被験者に負担軽減費が支払われない場合がある。
・ 医師主導治験では、検査・画像診断や、同種同効薬も保険外併用療養費の給付対象となる。
4|医師主導治験の場合、治験保険への加入が推奨される
医師主導治験では、自ら治験を実施しようとする者は、治験に関連して被験者に生じた健康被害に対する補償に備えて、保険への加入の措置、副作用等の治療に関する医療体制の提供等の措置を講じることとされている18。
医師主導治験保険は、補償金に対するもので、一定水準を超える健康被害(死亡または後遺障害)の救済のための準備として利用される。ただし、抗がん剤や免疫抑制剤などでは、疾患や重症度によっては、保険に加入できない場合もある。
なお、過失のある医療行為に起因して生じた健康被害は、医師賠償責任保険がカバーするため、医師主導治験保険では補償されない。このため、治験を実施する者(治験責任医師)は、医師賠償責任保険と医師主導治験保険の両方に加入しておくことが求められる19。
企業治験の場合には、治験薬の欠陥に伴う賠償事故に備えて、治験依頼者(医薬品メーカー)が、生産物賠償責任保険(PL保険)に加入していることが一般的である。
18 GCP省令第15条の9(被験者に対する補償措置)による。.
19 医師主導治験保険の補償責任条項の範囲は、損保会社により異なる。治験保険に加入した場合、補償の内容を被験者に説明するための「補償概要」を、自ら治験を実施しようとする者が作成することとなる。
6――安全性対策
1|副作用は、有害事象のうち因果関係が否定できないもの
「有害事象」(Adverse Event, AE)とは、医薬品を投与された被験者に生じた、全ての好ましくない、あるいは意図しない徴候、症状、疫病をいう。「副作用」(Adverse Drug Reaction, ADR)は、有害事象のうち、医薬品との因果関係が否定できないものをいう。
つぎの例で、AEとADRの違いをみてみよう。
・薬剤投与を受けた被験者が歩いていたら、自転車が衝突してきて、足を骨折した。この場合は、薬剤投与と衝突の因果関係が否定できるため、副作用とはならないが、有害事象には該当する。
・薬剤投与を受けた被験者が自転車に乗っていたら、ふらついて転倒して足を骨折した。この場合は、薬剤投与と転倒の因果関係が否定できない限り、副作用となる。無論、有害事象でもある。
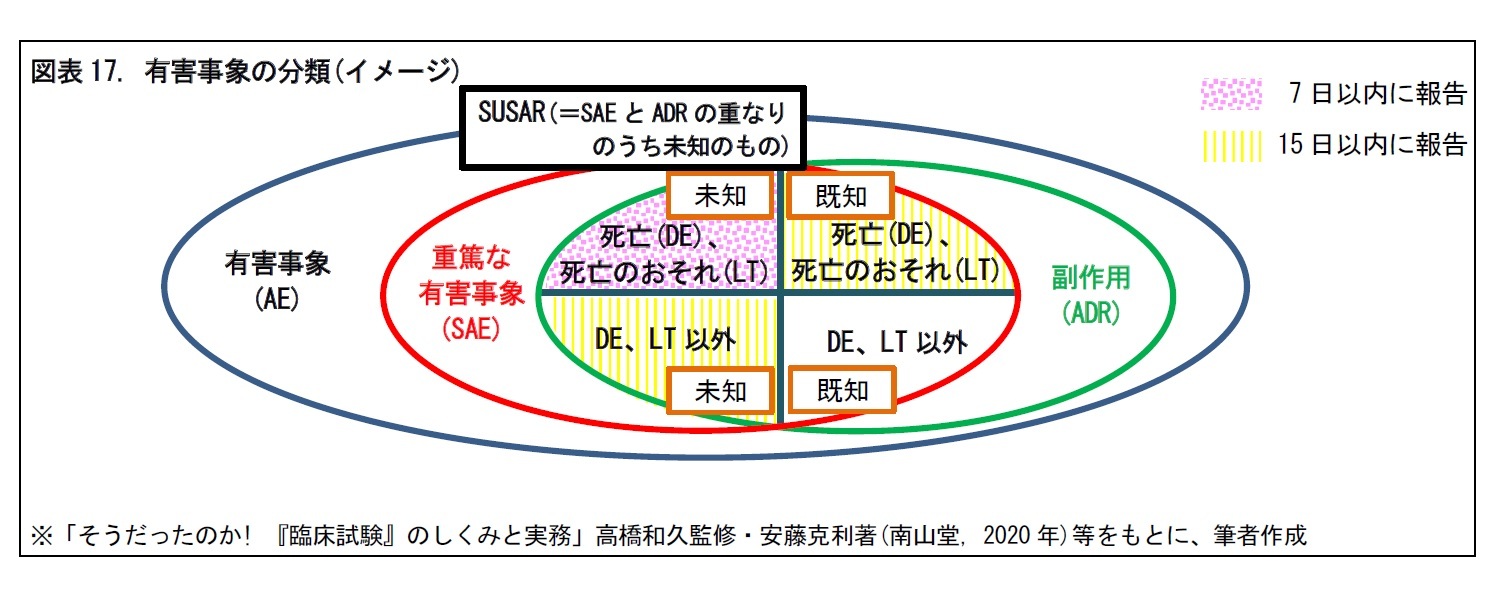
2|重篤な有害事象 (SAE) は、死亡、障害などを指す
重篤な有害事象(Serious Adverse Event, SAE)とは、有害事象のうち、つぎの①~⑦のいずれかに該当するものをいう。重篤性の判断には、死亡、障害、入院、先天異常が判断要素として用いられる。

![図表16. 重症度の判断基準 (CTCAEグレード)[抜粋]](/files/topics/68423_ext_15_31.jpg?site=nli)
20 CTCAEは、Common Terminology Criteria for Adverse Eventsの略で、有害事象共通用語規準を表す。CTCAEは、アメリカのNational Cancer Institute(NCI, 国立がん研究所)のCancer Therapy Evaluation Program(CTEP, がん治療評価研究プログラム)が公表しており、日本臨床腫瘍研究グループ(JCOG)によって和訳されている。
一般に、あるSAEの病状が副作用と判断された場合、既知(治験開始前より予測され、IBに記載されている)か、それとも、未知(IBに記載されていない)かの判定が必要となる。
SAEのうち、未知の副作用は、SUSAR(Suspected Unexpected Serious Adverse Reaction)と呼ばれる。SUSARのうち、死亡(DE)や死亡のおそれ(LT)の場合は7日以内、その他の場合は15日以内に規制当局に報告することが義務付けられている。一方、DEやLTの既知の副作用については、15日以内に規制当局に報告することが義務付けられている。
21 ただし、従前の例による治験届を提出したものには、被験薬のみを対象とする経過措置期間(2年間)が設けられている。
SAEが発生したときは、医師はただちに被験者に適切な緊急処置を行う。被験者に説明して、経緯をすべてカルテに記す。これに加えて、24時間以内に電話・FAXにより治験依頼者へ、書類により医療機関の長へ報告しなくてはならない。通常は、ただちに、まずCRAに連絡する。これは、「緊急報告」と呼ばれる第一報となる。SAE発生の連絡を受けたCRAは、治験責任医師・治験分担医師から詳細な情報を入手する。そのうえで、治験依頼者の安全性管理担当者に報告する。
その後、医師は、「重篤な有害事象に関する報告書」などの書類を作成し、速やかに治験依頼者と医療機関の長に報告する。これは、「詳細報告」と呼ばれる。CRAは、治験責任医師・治験分担医師のもとを訪問して、「重篤な有害事象に関する報告書」とカルテ等の原資料の整合性を確認する。
その上で、その報告書を治験依頼者に提出する。その報告においては、CRAが医師との面談により、「なぜSAEに気付いたのか ?」、「なぜその検査をしたのか ?」、「なぜ投与が延期・再開となったのか ?」など、評価において重要となる、原因に係る情報を把握しておくことが重視される。こうして詳細報告されたSAEは、治験審査委員会(IRB)で審議され、今後の治験継続の可否が判断される22。
治験依頼者は、SAEの発生を受けて、治験薬との因果関係や、予測可能性の評価を行い、PMDAへの報告の必要性を判断する。
ただし、一般に、有害事象は、治験薬との併用治療が原因で起こる場合や、もともとの病気(原疾患)が悪化して起こる場合、合併症が発生して起こる場合などがあり、明確な因果関係の特定や、因果関係の否定ができることは比較的まれとされる。特に、SAEの場合、通常は、発現頻度が少ないため、集団間の比較ではなく、個別症例ごとに医師が検討せざるを得ないといわれる。
22 医師主導治験で、複数の実施医療機関で治験が行われる場合、SAE発生時に医師が治験調整医師に報告する。報告を受けた治験調整医師は、SAEと治験薬との因果関係や、予測可能性の評価を行い、PMDAへの報告の必要性を判断する。
因果関係判定の考え方は、日本と欧米で異なっている。日本では、副作用は、「因果関係が否定できない有害事象」とされる。たとえば、医師がCRFを作成する際、CRFのフォーム上で、因果関係について、「関連あり」「関連があるかもしれない」「おそらく関連なし」「関連なし」の4つの選択肢から1つを選ぶ場合では、「関連なし」以外を選ぶと、すべて副作用として取り扱われる。
こうした日本の考え方は、副作用判定において極めて保守的である一方、報告される副作用が多くのノイズ(実は副作用ではないもの)を含むこととなり、シグナル(真の副作用)を検出できない可能性があるとの指摘もある。この考え方は、「Cannot be ruled out (否定できない)型」とされる。
一方、欧米では、「被験薬と有害事象の間に合理的な因果関係が説明できるもの」を「副作用の可能性があるもの」(Suspected Adverse Reaction, SAR)と定義して、報告対象としている。具体的には、「有害事象は、被験薬に関連しているという合理的な可能性はあるか?」との問いに対して、「はい(または不明)」であればSAR、「いいえ」であればSARではない、と判断する。
この欧米の考え方は、保守性よりも合理性を重視したものといえる。ただ、過去には、サリドマイド23や、エイズ24などの薬害問題も発生しており、合理性を重視することを危惧する声もある。この考え方は、「Reasonable possibility (合理的な可能性)型」と呼ばれる25。
こうした考え方の違いの結果、日本と欧米にまたがる国際共同治験を行うと、日本から報告される副作用には、欧米では報告対象とされていないものが含まれる、こととなる。
23 1950年代末~60年代初に、世界の40カ国以上で販売された鎮静・催眠薬「サリドマイド」による薬害事件。この薬を妊娠初期に服用すると、胎児の手/足/耳/内臓などに奇形を起こす。世界で数千人~1万人、日本で約千人の胎児が被害にあったと推定されている(死産を含む)。なお、アメリカはFDA(食品医薬品局)がこの薬剤を承認しなかった。アメリカのこのときの関係者は、後に、「アメリカ国民を薬害から守った」として、ケネディ大統領から表彰されている。
24 1980年代に、血友病患者の治療において、加熱処理によるウイルスの不活化を行わないまま、血液凝固因子製剤を使用したことにより、多数のHIV感染者およびエイズ患者を生み出した事件。薬害被害は世界的に起こったが、その中でもフランスや日本で被害が拡大し、医薬品メーカーの刑事責任を追及する結果となった。
23 アメリカは、従来「Cannot be ruled out型」を用いていたが、2011年9月に「Reasonable possibility型」に方針転換した。
治験依頼者は、治験中の被験薬について、有効成分ごとに、承認または開発中止となるまで、定期報告として、1年ごとに「治験安全性最新報告」(Development Safety Update Report, DSUR)を作成して、PMDAを通じて、厚生労働省に提出しなくてはならない。DSURには、医薬品の開発状況、1年間の調査期間中に得られたデータ・情報、それらに基づく分析・評価・結論が記載される。
また、2013年4月より、医療用医薬品とバイオ後続品の承認申請においては、医薬品リスク管理計画(Risk Management Plan, RMP)を提出することが求められるようになった。RMPは、大きく、「安全性検討事項」、「医薬品安全性監視計画」、「リスク最小化活動」の項目から構成される。
このうち、安全性検討事項は、リスク認識に関するもの。重要な特定されたリスク、重要な潜在リスク、重要な不足情報の洗い出しを行う。
医薬品安全性監視計画は、リスク情報の収集活動に関するもの。治験中の副作用症例の情報収集に加えて、市販直後調査、使用成績調査、製造販売後臨床試験によって情報収集が行われる。
リスク最小化活動は、リスクへの対応に関するもの。添付文書や患者向け医薬品ガイドにリスク情報を記載することに加えて、市販直後調査での情報提供、使用条件の各種設定などを通じて、リスクの最小化が図られる。
7――おわりに (私見)
本章では、まとめとして、治験について、筆者の私見を述べることとしたい。
〔1〕 医師主導治験のハードルを下げるべき
医師主導治験は、2010年代に入って増加傾向となっている。とはいえ、現在の治験の多くは、企業治験である。2020年度の医師主導治験の届出数は、治験全体の1割程度にとどまっている。一般に、小児疾患や希少疾患の場合、患者数が限られるため、医薬品メーカーにとって採算が合わず、治験が困難となる場合がある。こうした疾患では、医師主導治験への期待が大きい。
しかし、医師主導治験は、治験責任医師の業務負荷が過重となる。また、費用負担の面では、被験者に負担軽減費が支払われない場合がある。
こうした、医師主導治験のハードルを下げることで、医師が治験を行いやすく、また、患者が治験に参加しやすくなる仕組みづくりが欠かせない、と考えられる。
〔2〕 因果関係の判定基準のあり方について議論を始めるべき
新型コロナウイルス感染症の治療薬やワクチンの開発で、注目されたものの1つに国際共同治験がある。国際共同治験は、複数の国の治験データを共有して、効率的に承認申請・審査を行う取り組みである。その届出数は、2000年代に増加し、現在は、日本で行われる第III相試験の半数以上が、国際共同治験となっている。
しかし、日本と欧米の因果関係判定の基準が相違しているため、個別症例の副作用の報告内容が異なっている。日本では、集団データに基づいて因果関係を評価する環境は、十分に整備されていない。
まず、集団データを用いた因果関係評価を行うための環境作りが必要とみられる。そのうえで、判定基準のあり方について議論を開始し、国際的な創薬体制の向上につなげるべき、と考えられる。
〔3〕 人権保護や安全性対策の実効性を高めるべき
治験は、新薬や新たな治療法の承認を得ることを主たる目的として行われる。その際、被験者の人権や、安全性の確保を欠くことがあってはならない。そのために、被験者の候補者からの自由意思にもとづくインフォームド・コンセントの取得や、未知の副作用に該当する重篤な有害事象(SUSAR)の報告など、さまざまなルールや制度が整備されてきた。
しかし、ルールや制度をいくら充実させても、治験関係者の間で、人権保護や安全性対策の意識が乏しければ、こうした整備は画餅に帰す恐れがある。
新型コロナウイルス感染症の流行を受けて、治療薬・ワクチン開発に対する、社会的な期待は高まっている。それとともに、治験成功への圧力は増しているとみられる。こうした圧力に屈して、人権保護や安全性確保が損なわれることのないよう、治験関係者の意識向上を含めて、対策の実効性を高めることが必要、と考えられる。
【参考文献・資料】
(下記1~5の文献・資料は、包括的に参考にした)
- 「そうだったのか! 『臨床試験』のしくみと実務」高橋和久監修・安藤克利著(南山堂, 2020年)
- 「治験薬学(改訂第2版)-治験のプロセスとスタッフの役割と責任」亀井淳三・鈴木彰人編(南江堂, 2020年)
- 「医師主導治験START BOOK」内田英二編, 須崎友紀・川村芳江著(南山堂, 2016年)
- 「CRCのための治験業務マニュアル 第3版」亀山周二監修・CRCのための治験業務マニュアル作成委員会編(じほう, 2020年)
- 「徹底研究『治験』と『臨床』-運用の視点・患者の視点で読み解く」公益財団法人 医療科学研究所監修(法研, 2018年)
(下記の文献・資料は、内容の一部を参考にした)
- 「一般診療と治験の違い」(社会医療法人博愛会ホームページ, ライブラリー)
http://www.sagara.or.jp/libraries/index.php/01nyusen/727/ - 「医薬品の臨床試験の実施の基準に関する省令」(平成9年厚生省令第28号)
- 「薬物の治験計画届出件数の推移」(独立行政法人医薬品医療機器総合機構(PMDA))
- 「有害事象共通用語規準v5.0日本語訳JCOG版(CTCAE v5.0 - JCOG)」(日本臨床腫瘍研究グループ(JCOG))
- 「治験中副作用等報告数」(独立行政法人医薬品医療機器総合機構(PMDA))
- 「医薬品GCP省令改正箇所(2020年12月、2021年1月改正)」(日本QA研究会 GCP部会 特別プロジェクト3, 2021年4月15日)
(なお、下記2編の拙稿については、本稿執筆の基礎とした)
- 「医薬品・医療機器の現状(前編)-後発薬 (ジェネリック医薬品) への切り替えは、医療費削減の切り札となるのか? 」篠原拓也(ニッセイ基礎研究所 基礎研レポート, 2015年7月28日)
https://www.nli-research.co.jp/report/detail/id=42615?site=nli - 「医薬品・医療機器の現状(後編)-患者の残薬問題解消のために、かかりつけ薬局は何をすべきか?」篠原拓也(ニッセイ基礎研究所 基礎研レポート, 2015年8月3日)
https://www.nli-research.co.jp/report/detail/id=42630?site=nli

 各種レポート配信をメールでお知らせ。読み逃しを防ぎます!
各種レポート配信をメールでお知らせ。読み逃しを防ぎます!
保険研究部 主席研究員 兼 気候変動リサーチセンター チーフ気候変動アナリスト 兼 ヘルスケアリサーチセンター 主席研究員
篠原 拓也 (しのはら たくや)
研究・専門分野
保険商品・計理、共済計理人・コンサルティング業務
03-3512-1823
- 【職歴】
1992年 日本生命保険相互会社入社
2014年 ニッセイ基礎研究所へ
【加入団体等】
・日本アクチュアリー会 正会員
(2021年08月04日「基礎研レポート」)


公式SNSアカウント
新着レポートを随時お届け!日々の情報収集にぜひご活用ください。





新着記事
-
2024年04月24日
中国経済の現状と注目点-24年1~3月期は好調な出だしとなるも、勢いが持続するかは疑問 -
2024年04月24日
人手不足とインフレ・賃上げを考える -
2024年04月24日
米国でのiPhone競争法訴訟-司法省等が違法な独占確保につき訴え -
2024年04月23日
他国との再保険の監督に関する留意事項の検討(欧州)-EIOPAの声明 -
2024年04月23日
気候変動-温暖化の情報提示-気候変動問題の科学の専門家は“ドラマが少ない方向に誤る?”
レポート紹介
-
研究領域
-
経済
-
金融・為替
-
資産運用・資産形成
-
年金
-
社会保障制度
-
保険
-
不動産
-
経営・ビジネス
-
暮らし
-
ジェロントロジー(高齢社会総合研究)
-
医療・介護・健康・ヘルスケア
-
政策提言
-
-
注目テーマ・キーワード
-
統計・指標・重要イベント
-
媒体
- アクセスランキング
お知らせ
-
2024年04月02日
News Release
-
2024年02月19日
News Release
-
2023年07月03日
News Release
【治験の実務-臨床試験の現状 (後編)】【シンクタンク】ニッセイ基礎研究所は、保険・年金・社会保障、経済・金融・不動産、暮らし・高齢社会、経営・ビジネスなどの各専門領域の研究員を抱え、様々な情報提供を行っています。
治験の実務-臨床試験の現状 (後編)のレポート Topへ- 新型コロナウイルス
- ウィズコロナ・アフターコロナ
- 生成AI・AI
- IoT
- デジタルトランスフォーメーション(DX)
- フィンテック(FinTech)
- キャッシュレス
- デジタル通貨
- デジタルプラットフォーム
- マイナンバー
- MaaS、CASE
- SDGs
- ESG
- 気候変動
- カーボンニュートラル・脱炭素社会
- 経済安全保障
- 供給網(サプライ・チェーン)
- イデコ(iDeCo)
- 新NISA・NISA
- 確定拠出年金
- 企業型DC
- 資産所得倍増プラン
- 日本銀行
- 人手不足・人材不足
- 働き方改革
- テレワーク・在宅勤務
- ダイバーシティ(多様性)社会
- 外国人雇用・就労
- 地域包括ケアシステム
- 認知症
- 金融(ファイナンシャル)ジェロントロジー
- 全世代型社会保障会議
- 年金制度改革
- 社会保障・税改革
- 医療・介護制度改革
- 健康寿命
- 健康経営
- 格差・貧困
- 世代間格差
- 未婚・晩婚・非婚
- 少子高齢化
- シェアリングエコノミー
- Z世代
- オフィスレントインデックス
- 生命保険事業概況
- ブレグジット(Brexit・イギリスEU離脱)
- 米中貿易摩擦
- 米国
- 中国
- 欧州
- アジア・新興国
- 韓国
- ASEAN
- インド
- 統計
- 消費者物価指数(CPI)│日本
- 雇用統計│日本
- 鉱工業生産指数│日本
- 貿易統計│日本
- 法人企業統計│日本
- QE速報・予測
- 日銀金融政策決定会合
- 日銀短観│日本
- 資金循環統計│日本
- 景気ウォッチャー調査│日本
- 地域経済報告(さくらレポート)
- マネタリーベース│日本
- GDP等│米国
- FOMC(連邦公開市場委員会)│米国
- 住宅販売・着工│米国
- 雇用統計│米国
- 米個人所得・支出|米国
- ECB政策理事会│欧州
- ユーロ圏消費者物価指数
- ユーロ圏GDP
- ユーロ圏失業率
- 英国雇用関連統計
- 英国金融政策
- 英国GDP
- 将来人口推計
- 人口動態統計
- 宿泊旅行統計
- 貿易統計|ASEAN
- インドGDP
- インド消費者物価
- タイGDP
- マレーシアGDP
- フィリピンGDP
- インドネシアGDP
- ロシアGDP
- ブラジルGDP
- IMF世界経済見通し
- 企業物価指数
Copyright © NLI Research Institute. All rights reserved.